Case Report: IgM-Kappa Restricted AL Amyloidosis with Renal Involvement

AL amyloidosis is a rare plasma cell dyscrasia characterized by the deposition of misfolded light chains in various organs. We report a case of a 38-year-old male with recurrent bilateral leg swelling and proteinuria, diagnosed with renal AL amyloidosis with IgM-kappa restriction. The diagnosis was confirmed via renal biopsy, serum free light chain assay, and immunohistochemical studies. The patient was initiated on bortezomib-based therapy. This case highlights the importance of early recognition and targeted treatment of AL amyloidosis, especially in the context of IgM paraproteinemia.
Introduction
AL (light-chain) amyloidosis is a systemic disorder caused by the deposition of monoclonal immunoglobulin light chains, primarily produced by clonal plasma cells. The disease most commonly affects the kidneys, heart, and nervous system, leading to progressive organ dysfunction. IgM-associated AL amyloidosis is rare and often linked to lymphoproliferative disorders, including Waldenström’s macroglobulinemia or IgM myeloma. Early diagnosis and appropriate treatment can significantly impact prognosis.
Case Presentation
Clinical History
A 34-year-old male, known to have Diabetes Mellitus (DM), Hypertension (HTN), Bronchial Asthma (BA), and HbE trait, presented with recurrent bilateral leg swelling, fatigue and progressive weakness over several weeks.
Physical Examination
BP: 130/85 mmHg
Pulse: 88 bpm, regular
No hepatosplenomegaly or lymphadenopathy
Bilateral pitting edema present up to the mid-shins
Laboratory Investigations
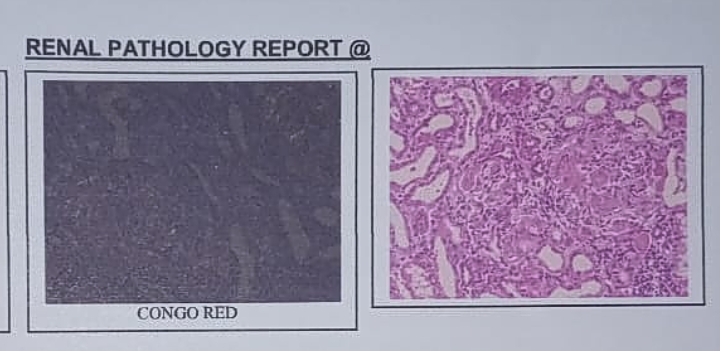
Renal Biopsy Findings
Histopathology:
Mesangial expansion with eosinophilic deposits
Positive Congo red staining with apple-green birefringence under polarized light
Interstitial fibrosis and tubular atrophy (20%)
Immunofluorescence & DIF (Direct Immunofluorescence):
IgM & Kappa Light Chain Positive
Lambda Light Chain Negative
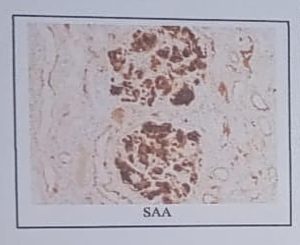
Immunohistochemistry:
Positive for Serum Amyloid A (SAA)
Diagnosis
Renal AL amyloidosis with IgM-kappa restriction, likely associated with an underlying plasma cell disorder.
Management
Hematological Workup & Risk Stratification
Bone Marrow Aspiration & Biopsy: Clonal plasma cell infiltration
Serum & Urine Immunofixation Electrophoresis: IgM-Kappa monoclonal protein
MYD88 Mutation Analysis: Negative (suggesting IgM myeloma rather than Waldenström’s macroglobulinemia)
Cardiac Workup: NT-proBNP & Troponin-I normal
Treatment Plan
First-line Therapy (BDR Regimen):
- Bortezomib (1.3 mg/m² SC, weekly)
- Dexamethasone (20 mg/week)
- Rituximab (375 mg/m² IV, if Waldenström’s suspected)
Supportive Therapy:
- Renal protection: RAAS inhibitors (if tolerated)
- Diuretics: For fluid overload
- Neuropathy management: Gabapentin/Pregabalin
- Nutritional support
Transplant Consideration:
Autologous stem cell transplantation (ASCT) evaluated, but deferred due to renal dysfunction
Follow-up and Prognosis
The patient showed partial hematological response after 3 cycles, with a reduction in proteinuria and stabilization of renal function. Ongoing follow-up with repeat serum free light chains and NT-proBNP is planned.
Discussion
AL amyloidosis with IgM paraproteinemia is rare, comprising <5% of all AL cases. The differential diagnosis includes IgM myeloma, Waldenström’s macroglobulinemia, and monoclonal gammopathy of renal significance (MGRS). The treatment approach differs from standard AL amyloidosis due to the unique biology of IgM-secreting clones. Bortezomib-based regimens have shown efficacy in such cases, especially when ASCT is not feasible.
Early recognition is crucial, as renal involvement is associated with poor prognosis if untreated. Regular monitoring for hematologic and organ response is essential for optimizing treatment.
Conclusion
This case highlights IgM-kappa AL amyloidosis with significant renal involvement, managed successfully with bortezomib-based therapy. The disease requires a multidisciplinary approach, with careful differentiation from Waldenström’s macroglobulinemia and MGRS.
Key Takeaways:
- IgM-associated AL amyloidosis is rare and requires differentiation from Waldenström’s macroglobulinemia.
- Renal involvement is common, with proteinuria being a key clinical feature.
- Bortezomib-based therapy is the first-line treatment, and ASCT should be considered if feasible.
- Close monitoring of hematologic and organ response is crucial for prognosis.
References
1. Merlini G, Palladini G. Amyloidosis: is a cure possible? Ann Oncol. 2008;19 Suppl 4:iv63-6.
2. Wechalekar AD, Gillmore JD, Hawkins PN. Systemic amyloidosis. Lancet. 2016;387(10038):2641-54.
3. Palladini G, Schönland SO, Sanchorawala V, et al. Systemic light chain amyloidosis: an update for treating physicians. Blood Cancer J. 2020;10(7):71.
4. Kumar S, Dispenzieri A, Lacy MQ, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. 2012;30(9):989-95.
5. Palladini G, Kastritis E, Maurer MS, et al. Daratumumab-based treatment for amyloid light-chain amyloidosis. N Engl J Med. 2021;385(1):46-58.
6. Sanchorawala V. AL amyloidosis: advancements in diagnosis and treatment. Best Pract Res Clin Haematol. 2020;33(1):101138.
7. Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995;32(1):45-59.