Abstract
Hemoglobin H (Hb H) disease is a moderate to severe form of alpha thalassemia resulting from impaired alpha globin production. We report a case of a patient with Hb H disease caused by compound heterozygosity for a deletional -3.7 mutation and a Hb Constant Spring (Hb CS) mutation. The patient presented with chronic anemia and became transfusion-dependent at 14 months of age. This case highlights the clinical manifestations, molecular genetics, and management challenges associated with Hb H disease.
Introduction
Alpha thalassemia results from deletions or mutations in the alpha globin genes, leading to deficient production of alpha globin chains in hemoglobin. Hb H disease is a clinically significant form of alpha thalassemia, characterized by the formation of abnormal hemoglobin (β4 tetramers) due to a marked reduction in alpha globin synthesis. Patients with Hb H disease can exhibit variable clinical severity, depending on the specific genetic mutations involved. The presence of a Hb Constant Spring mutation, a non-deletional alpha thalassemia variant, often exacerbates disease severity.
Case Presentation
A 2.5 year boy child was diagnosed with Hb H disease following persistent anemia and hepatosplenomegaly detected during infancy. The patient was born to non-consanguineous parents and had no known family history of thalassemia.
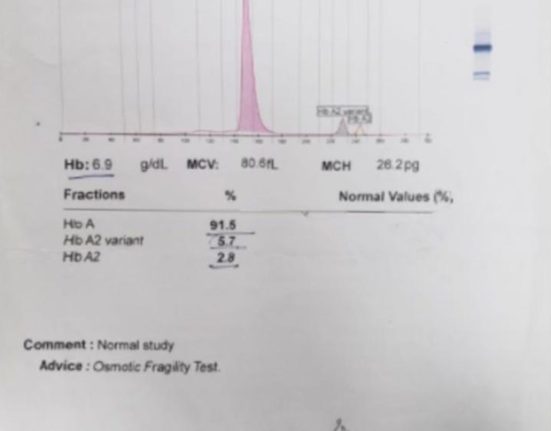
At 14 months of age, the patient developed worsening pallor, fatigue, and poor weight gain, necessitating regular blood transfusions. Laboratory investigations revealed microcytic hypochromic anemia with hemoglobin levels ranging from [insert Hb levels] g/dL before transfusion. Hemoglobin electrophoresis demonstrated an elevated percentage of Hb H, and genetic analysis confirmed a compound heterozygous state with a deletional -3.7 mutation and a Hb Constant Spring mutation.
Discussion
Molecular Pathogenesis
The -3.7 deletion is a common form of single alpha globin gene deletion, leading to decreased alpha globin synthesis. The Hb Constant Spring mutation results from a point mutation in the stop codon of the alpha2-globin gene, producing an unstable and elongated alpha globin chain. The combination of these mutations significantly impairs alpha globin production, leading to an excess of beta chains, which aggregate into Hb H (β4), an unstable hemoglobin variant.
Clinical Manifestations and Management
Patients with Hb H disease generally present with mild to moderate anemia, jaundice, hepatosplenomegaly, and, in severe cases, transfusion dependency. This patient exhibited early-onset transfusion dependence, indicating a more severe phenotype, similar to non-deletional Hb H disease or alpha thalassemia major.
Long-term transfusion therapy poses risks, including iron overload, necessitating chelation therapy to prevent complications such as cardiac dysfunction and endocrinopathies. The patient was started on oral Iron chelation therapy to manage iron overload. Other supportive measures included folic acid supplementation and regular monitoring for hemolysis and splenomegaly progression.
Prognosis and Future Considerations
While many patients with Hb H disease can maintain adequate hemoglobin levels without transfusions, those with Hb Constant Spring mutations often have a more severe disease course. The role of hematopoietic stem cell transplantation (HSCT) as a curative option remains under investigation for severe cases. Genetic counseling was provided to the family, highlighting reproductive risks and prenatal diagnostic options.
Conclusion
This case of Hb H disease with a compound heterozygous -3.7 deletion and Hb Constant Spring mutation underscores the genetic complexity and variable clinical spectrum of alpha thalassemia. Early diagnosis, regular transfusion support, and iron chelation therapy are crucial for improving patient outcomes. Further research is needed to explore novel therapeutic strategies for severe forms of alpha thalassemia.
Acknowledgments
My beloved patient Master X.
Conflict of Interest
No conflict of interest.